Midterm test simulation¶
Before you start¶
Please write one single python script.
IMPORTANT: Add your name and ID (matricola) on top of each .py file!
Problem 1¶
Given a text string \(S\) (possibly on several lines), write a python function that takes the string \(S\) and a second string \(S_1\) (note: assumed to be shorter than S) in inpu and returns a list with all positions at which \(S_1\) starts in \(S\). If no such positions exist a warning message should be printed.
Ex.
S = """TGAATACATATTATTCCGC
GTCCTACCAAAAAATATACATAGATAT
GATACTAGGGGACCGGTTCCGGGATGATGAT"""
print(findPositions(S, "TGA"))
print(findPositions(S, "GAT"))
print(findPositions(S, "none"))
should return:
String 'TGA' found at the following positions:
[0, 45, 70, 73]
String 'GAT' found at the following positions:
[41, 46, 68, 71, 74]
Warning: string 'none' not found. Returning None
None
Problem 2¶
The .tsv file gene_models.tsv is a compact representation of the
gene models on the genome having sequence specified in
sequences.fasta. The first few lines are reported below:
Chr feature start end ID
Chr05 gene 50988 51101 gene:MD05G1000100
Chr05 ncRNA 50988 51101 ncRNA:MD05G1000100
Chr05 gene 83210 83329 gene:MD05G1000200
Chr05 ncRNA 83210 83329 ncRNA:MD05G1000200
Chr05 gene 87650 91333 gene:MD05G1000300
Chr05 mRNA 87650 91333 mRNA:MD05G1000300
Chr05 CDS 87650 87727 CDS:MD05G1000300.8
as the header (first line) describes, the first column is the chromosome containing the feature (that can be gene, ncRNA, exon, CDS, …), the third and fourth columns are the start and end position of the feature on the chromosome and the fifth row is the identifier of the feature ID. Note that, unlike python strings, features in ``gene_models`` start from coordinate 1 instead of 0.
The file sequences.fasta stores sequence information in .fasta
format. A mock entry is the following:
>Chr01
AGGCCTAGGTCTTCCAGAGTCGCTTTTTCCAGCTCCAGACCGATCTCTTCAG
AGGCCAATCGCCAGTTTACCACATACACCCAGACCGATCTCTTCAG
where the first line is the identifier of the read and starts with a “>”. The sequence follows the line with the identifier and can be on multiple lines.
Implement the following python functions:
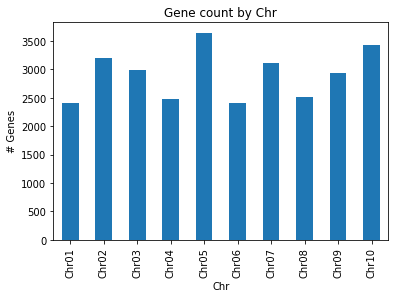
computeGeneStats(filename): gets thefilenameof a .tsv file as explained above, stores its content in a suitable data structure of your choice (hint: pandas might help here), counts (and prints) the number of features of type gene, printing the minimum, maximum and average gene length in the whole file. The function should also produce a bar plot of the number of genes per chromosome.
Note: The function should return the data structure containing all the data.
Calling:
fn = "gene_models.tsv"
seqFile = "sequences.fasta"
GenesDF = computeGeneStats(fn)
should give:
29136 genes present
Avg gene length: 2724.71
Min gene length: 45
Max gene length: 53052
printSequence(geneInfo, geneID, sequenceFile): gets thegeneInfodata structure created by computeGeneStats, a gene identifiergeneIDand the filename of a fasta formatted filesequenceFileand ifgeneIDis present ingeneInfoit prints its length and a fasta-formatted string reporting the sequence of the gene Note that chromosome, start and end position are saved ingeneInfo. If the chromosome is not in the sequenceFile or geneID is not in geneInfo the function should display a corresponding message (see examples below). Optional: add a parameteroutFilewith default value None, that if set (it will contain the path to an output file) makes the function write the fasta entries to fileoutFilerather than to the screen.
Hint: you can use biophtyon to read a fasta file.
For example, assuming an entry:
ChrX gene 3 7 gene:geneid1
in geneInfo and a sequence:
>ChrX
GATTACATAACACACTACA
in sequenceFile, calling
printSequence(geneInfo,"geneid1",sequenceFile) should print:
Gene geneid1 is in ChrX and has length 5
>geneid1:ChrX:3_7
TTACA
Given GenesDF as returned by computeGeneStats, calling:
printSequence(GenesDF,"MD05G1000100",seqFile)
print("")
printSequence(GenesDF,"MD03G1000400",seqFile)
print("")
printSequence(GenesDF,"MD08G1000100",seqFile)
print("")
printSequence(GenesDF,"MD08G100019191",seqFile)
should give:
Gene MD05G1000100 is in Chr05 and has length 114
>MD05G1000100:Chr05:50988_51101
CGCCCAAGGGAGTGGATCCTGTAAGCCTTATATGTATATTCCCATCTCTACCTAGCACGAGGCCTTTTGGGAGCTCATTGGCTTCGGGTTATTGTCACATCCCGACCCGAGCCC
Gene MD03G1000400 is in Chr03 and has length 189
>MD03G1000400:Chr03:21801_21989
CGTATTATAGGAATTCTTCTTCCATTGAAAGGATGGATTGAAAGGATGGATGGCATGTTGTCTATACCTCTCGTCATTAGGATGTACAATAATGGCAGTATCACCGAGCATAGTTTCCACTCTAGTGGTGGCCACCATAATCTCACCAAAGTCTTCCTCTAGAGTATAAGCAAATGAAGTTAACATGCC
Gene MD08G1000100 is in Chr08 and has length 5384
Chr08 is not present in the sequence file.
GeneID MD08G100019191 is not present in the input file.
Gene MD05G1000100 is in Chr05 and has length 114
- (Optional) Modify the function
computeGeneStats(filename)to make it also plot a boxplot of the gene lengths per chromosome.
Expected output: 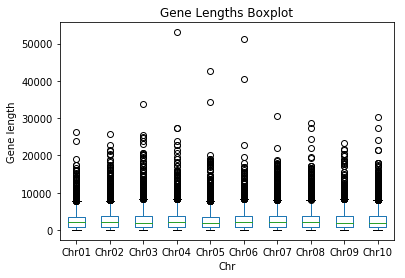
Available material for exams¶
The course notes can be used as the slides of the theory and practicals. Documents on the libraries are here:
You can download all of them here: archive
Download the data¶
Create a “qcbsciprolab-2019-31-10-NAME-SURNAME-ID” folder on the desktop. Download the following data to the folder that you just created.
The gene models .tsv file: gene_models.tsv and the fasta file with the sequences: sequences.fasta. Note that the files are gzipped, so before using them gunzip them in a terminal with:
gunzip sequences.fasta.gz
gunzip gene_models.tsv.gz
A possible solution:¶
Problem 1¶
In [1]:
%reset -s -f
def findPositions(S, S1):
s1len = len(S1)
S = S.replace("\n","")
ret = [x for x in range(0,len(S)) if S[x:x+s1len] == S1]
if len(ret) > 0:
print("\nString '{}' found at the following positions:".format(S1))
return ret
else:
print("\nWarning: string '{}' not found. Returning None".format(S1,S))
S = """TGAATACATATTATTCCGC
GTCCTACCAAAAAATATACATAGATAT
GATACTAGGGGACCGGTTCCGGGATGATGAT"""
print(findPositions(S, "TGA"))
print(findPositions(S, "GAT"))
print(findPositions(S, "none"))
String 'TGA' found at the following positions:
[0, 45, 70, 73]
String 'GAT' found at the following positions:
[41, 46, 68, 71, 74]
Warning: string 'none' not found. Returning None
None
Problem 2¶
In [3]:
%reset -s -f
"""exercise.py"""
import matplotlib.pyplot as plt
import pandas as pd
import numpy as np
from Bio import SeqIO
def computeGeneStats(filename):
geneInfo = pd.read_csv(filename, sep="\t", header = 0)
genes = geneInfo[ geneInfo["feature"] == "gene"]
#print(genes.head())
print("{} genes present".format(genes.shape[0]))
#print(genes.head())
avg = np.mean(genes["end"] - genes["start"] + 1)
m = np.min(genes["end"] - genes["start"] + 1 )
M = np.max(genes["end"] - genes["start"] + 1 )
print("Avg gene length: {:.2f}\nMin gene length: {}\nMax gene length: {}".format(avg, m, M))
grpd = genes.groupby(["Chr"])
agg = grpd.aggregate(pd.DataFrame.count)["feature"]
agg.plot(kind = 'bar')
plt.title("Gene count by Chr")
plt.ylabel("# Genes")
plt.xlabel("Chr")
plt.show()
bpdata = {}
for i, g in grpd:
if(i not in bpdata):
bpdata[i] = []
bpdata[i] = g["end"] - g["start"] + 1
pdData = pd.DataFrame(bpdata)
pdData.plot(kind='box')
plt.title("Gene Lengths Boxplot")
plt.ylabel("Gene length")
plt.xlabel("Chr")
plt.show()
return geneInfo
def printSequence(geneInfo, geneID, sequenceFile, outFile = None):
gn = "gene:" + geneID
gene = geneInfo[geneInfo["ID"] == gn]
of = None
if outFile != None:
of = open(outFile, "w")
if len(gene) > 0:
seqDict = SeqIO.to_dict(SeqIO.parse(sequenceFile, "fasta"))
chr = gene.iloc[0]["Chr"]
start = gene.iloc[0]["start"]
end = gene.iloc[0]["end"]
print("Gene {} is in {} and has length {}".format(geneID,chr, end-start + 1))
if chr in seqDict:
s = seqDict[chr].seq[start-1:end ]
if outFile != None:
of.write(">"+geneID + ":" + chr +":" + str(start) + "_" + str(end)+"\n")
of.write(str(s)+"\n")
else:
print(">"+geneID + ":" + chr +":" + str(start) + "_" + str(end))
print(str(s))
else:
print(chr, "is not present in the sequence file.")
else:
print("GeneID {} is not present in the input file.".format(geneID))
if outFile != None:
of.close()
fn = "./midterm_sim/gene_models.tsv"
seqFile = "./midterm_sim/sequences.fasta"
GenesDF = computeGeneStats(fn)
printSequence(GenesDF,"MD05G1000100",seqFile)
print("")
printSequence(GenesDF,"MD03G1000400",seqFile)
print("")
printSequence(GenesDF,"MD08G1000100",seqFile)
print("")
printSequence(GenesDF,"MD08G100019191",seqFile)
printSequence(GenesDF,"MD05G1000100",seqFile, "MD05G1000100.fasta")
29136 genes present
Avg gene length: 2724.71
Min gene length: 45
Max gene length: 53052
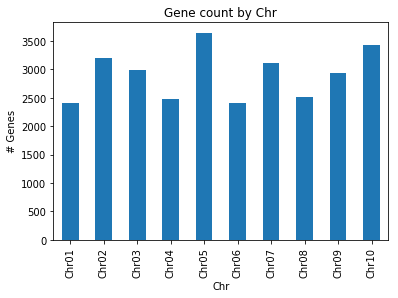
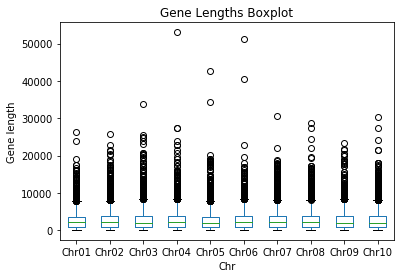
Gene MD05G1000100 is in Chr05 and has length 114
>MD05G1000100:Chr05:50988_51101
CGCCCAAGGGAGTGGATCCTGTAAGCCTTATATGTATATTCCCATCTCTACCTAGCACGAGGCCTTTTGGGAGCTCATTGGCTTCGGGTTATTGTCACATCCCGACCCGAGCCC
Gene MD03G1000400 is in Chr03 and has length 189
>MD03G1000400:Chr03:21801_21989
CGTATTATAGGAATTCTTCTTCCATTGAAAGGATGGATTGAAAGGATGGATGGCATGTTGTCTATACCTCTCGTCATTAGGATGTACAATAATGGCAGTATCACCGAGCATAGTTTCCACTCTAGTGGTGGCCACCATAATCTCACCAAAGTCTTCCTCTAGAGTATAAGCAAATGAAGTTAACATGCC
Gene MD08G1000100 is in Chr08 and has length 5384
Chr08 is not present in the sequence file.
GeneID MD08G100019191 is not present in the input file.
Gene MD05G1000100 is in Chr05 and has length 114